
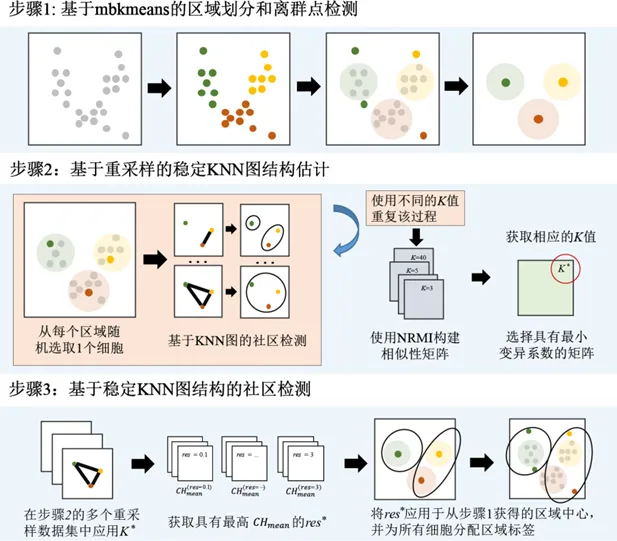
随着单细胞RNA测序技术在生命、医学领域的快速普及,单细胞数据规模日益庞大。传统的单细胞聚类分析面临着一致性差、时间效率低和计算复杂度高等诸多问题。尤其是在处理多批次、不平衡数据集时,分群结果不稳定影响了很多师生的课题进展。针对上述问题,厦门大学医学院统计支持办公室近期上线了一种全新的超大规模单细胞转录组分析框架,CDSKNNXMBD[1],通过优化的稳定KNN图结构,为单细胞聚类分析提供了一种高效且准确的解决方案。 一、CDSKNNXMBD框架原理和优势 适用于大规模数据集:有效解决大规模数据集带来的计算挑战。2. 优化细胞数目不平衡:改善了数据不平衡下的聚类分析。3. 基于优化的稳定图结构,有效避免了批次效应和混肴信息对聚类的影响。
二、性能对比 我们将CDSKNNXMBD与其他主流聚类方法进行了比较,包括PARC[2],phenograph[3],FlowGrid[4]。对比角度包括聚类精度(ARI),分簇个数和细胞类型个数的偏差,以及算法的运行时长:在人类胎儿图谱的单细胞数据集上,对比结果显示CDSKNN XMBD在处理数据不平衡情况下表现出色,能够更准确地估计细胞类型的数量,相比其他方法具有更高的聚类一致性;
在不同生物背景的数据集上,CDSKNNXMBD表现出很好的适应性和稳定性,CDSKNNXMBD在处理大规模单细胞数据时表现出更高的运行效率,相比其他方法能够显著节省时间,尤其在处理百万级单细胞数据时表现突出。值得一提的是,在处理三个百万级单细胞数据集时,CDSKNNXMBD时间消耗的中位数水平仅为6.18-8.22分钟,相较于其他三种方法,其运行时间减少了33.3%至99%。FlowGrid在处理146万个数据时的平均运行时间达到了1403分钟,而CDSKNNXMBD仅需6.33分钟。
我们对不同主成分个数下的聚类性能进行了比较,发现CDSKNNXMBD在不同主成分个数下都能够保持较高的聚类一致性和稳定性。具体来说,随着主成分个数的增加,CDSKNNXMBD的聚类性能并未出现明显下降,而且在保持较高聚类一致性的同时,计算时长也保持相对稳定。相比之下,其他方法在主成分个数增加时可能出现聚类性能下降和计算时长增加的情况。
综合来看,CDSKNNXMBD是一种灵活、高效的聚类工具,特别适用于处理不平衡数据和大规模单细胞转录组数据。 自助分析流程 研究者可以在R环境下方便的构建自己的CDSKNNXMBD标准分析流程: (1)R包安装 1. install.packages("devtools") 2. devtools::install_github("renjun0324/CDSKNN") (2)快速开始 1. data(pca_result) 2. data(cellinfo) 3. 4. result = CDSKNNXMBD(pca_result, 5. outlier_q = 0.1, 6. down_n = 300, 7. knn_range = 5:70, 8. iter = 100, 9. compute_index = c("Davies_Bouldin","Calinski_Harabasz"), 10. assess_index = "Davies_Bouldin", 11. cores = 1, 12. seed = 723) 13. 14. # ARI result 15. library(aricode) 16. ARI(result$cluster_df$cluster,cellinfo$celltype) (3)分步骤进行 1. # outlier detect 2. outlier_kmeans = OutlierKmeans(dataMatrix = pca_result, 3. outlier_q = 0.1, 4. down_n = 300, 5. cores = 1, 6. seed = 723) 7. 8. # random sampling and choose optmial KKN graph structure 9. sampling_result = SamplingLouvain(dataMatrix = pca_result, 10. outlier_kmeans = outlier_kmeans, 11. knn_range = 5:70, 12. iter = 100, 13. compute_index = c("Davies_Bouldin"), 14. cores = 10, 15. seed = 723) 16. 17. # get final clustering result 18. new_louvain = NewLouvain(sampling_result = sampling_result, 19. outlier_kmeans = outlier_kmeans, 20. assess_index = "Davies_Bouldin", 21. cores = 1) 22. 23. result = data.frame(row.names = rownames(new_louvain$cluster_meta), 24. name = rownames(new_louvain$cluster_meta), 25. celltype = cellinfo$celltype, 26. cluster = as.factor(new_louvain$cluster_meta$cluster), 27. stringsAsFactors = FALSE) 28. 29. # ARI result 30. library(aricode) 31. ARI(result$cluster_df[rownames(cellinfo),"cluster"],cellinfo$celltype) CDSKNNXMBD的引用信息请参考: Ren J, Lyu X, Guo J, et al. CDSKNNXMBD: a novel clustering framework for large-scale single-cell data based on a stable graph structure[J]. Journal of Translational Medicine, 2024, 22(1): 233. 厦门大学医学院统计支持办公室新实验方法咨询 厦门大学医学院统计支持办公室提供基于CDSKNNXMBD框架的定制化单细胞转录组分析流程,主要服务内容包括: (1)数据分析:包含实验数据整理、统计分析、高通量测序分析、图表设计及绘制和相关解释。 (2)咨询服务:提供统计方法、图表设计、样本量计算咨询。 (3)分析报告:包括数据分析结果总结和科研论文方法部分的撰写。 我们十分乐意为老师和同学们解答CDSKNNXMBD分析过程中的各种问题。无论是在数据处理、分析方法选择还是科研论文撰写阶段,我们都愿意为同学们提供专业的建议和支持! 地点:医学院健康医疗大数据国家研究院,爱礼楼201 联系电话:18030257072 邮箱:trans_medxmu@126.com 参考文献: [1] Ren J, Lyu X, Guo J, et al. CDSKNNXMBD: a novel clustering framework for large-scale single-cell data based on a stable graph structure[J]. Journal of Translational Medicine, 2024, 22(1): 233. [2] Stassen S V, Siu D M D, Lee K C M, et al. PARC: Ultrafast and accurate clustering of phenotypic data of millions of single cells[J]. Bioinformatics, 2020, 36(9): 2778-2786. [3] Levine J H, Simonds E F, Bendall S C, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis[J]. Cell, 2015, 162(1): 184-197. [4] Fang X, Ho J W K. FlowGrid enables fast clustering of very large single-cell RNA-seq data[J]. Bioinformatics, 2021, 38(1): 282-283. |





